Si ya hemos practicado un poco con las secuencias anteriores, deberíamos entender que una secuencia de nucleótidos precisa tiene un gran valor identificativo. Por ello, podemos deducir que gran parte del manejo del programa (Clonemanger) se basa en buscar secuencias concretas, alinearlas de manera parecida, etc. Y esas son herramientas que nos ayudarán a definir y entender cómo se logra encontrar un sistema CRISPR en una secuencia de nucleótidos, que como todos sabemos se trata de:
“Clustered Regularly Interspaced Short Palindromic Repeats”
O sea, grupos de secuencias cortas (20-40 nt) repetidas generalmente palindrómicas interespaciadas de manera regular (a distancias entre 30-70 nt).

Recordando, atendiendo y practicando las explicaciones del pasado Jueves 5 de Diciembre
Para identificar de manera rápida este tipo de “estructuras” en una secuencia de DNA existe una página web denominada: crispr finder (https://crispr.i2bc.paris-saclay.fr/Server/). Os animo a que entréis:

Y subáis el documento que tenéis en la carpeta 2-E coli denominado E coli.txt (que “pesa” 4596 KB). Y “clickeis” en Find crispr
Os debe de salir algo así:
Os debe de salir algo así:

Que debemos saber (y recordar) qué significan:

Con esta información deberíamos de saber, crear y modificar un documento de clone manager. De tal manera que partiendo de:
Una secuencia de este tipo:
Una secuencia de este tipo:

La convirtamos inicialmente en una secuencia de este tipo:

Para ello habrá que utilizar la herramienta add feature seleccionando en cada caso la secuencia que queremos “dibujar”. En este caso deberéis de ayudar a los que no estuvieron, y hacer un poco de memoria de cómo lo hicimos ese Jueves, clave!!! … (consultar el tutorial del programa por si sirve de ayuda, usad el blog, cargarlo de preguntas, etc… ).

Finalmente, acompañando a estos sistemas repetidos, casi siempre se encuentran grupos de genes asociados, los denominados genes “Cas”. El clone manager nos va a permitir identificarlos, “dibujarlos”, definirlos y en resumen entender en qué consisten.
Se trata de que debemos de convertir esta información:
Se trata de que debemos de convertir esta información:

En esta otra:
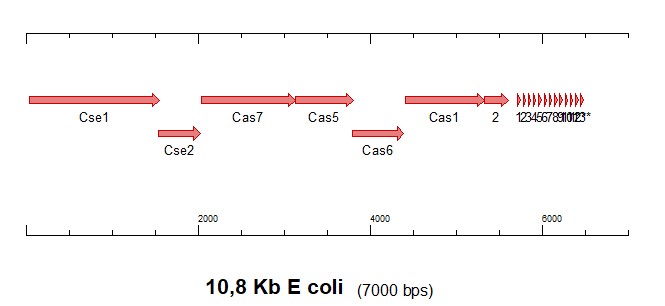
Donde se identifican los marcos abiertos de lectura (ORFs) que codifican las proteínas que acompañan el elemento CRISPR (con 13 repeticiones directas) y que hacen funcional todo un sistema de defensa inmune en bacterias.
Es sólo (y no tan sólo, soy consciente) de ponernos un rato y “entender” toda la información extraíble de una mera secuencia de cuatro letras: la A, la T, la C y la G… .
Ánimo que veréis como podéis (podemos). Activemos el blog con comentarios/preguntas, que estoy seguro que no sólo hay una sino mil….
Ya tenemos tarea!!!!...
Es sólo (y no tan sólo, soy consciente) de ponernos un rato y “entender” toda la información extraíble de una mera secuencia de cuatro letras: la A, la T, la C y la G… .
Ánimo que veréis como podéis (podemos). Activemos el blog con comentarios/preguntas, que estoy seguro que no sólo hay una sino mil….
Ya tenemos tarea!!!!...











